
Prevalentie homozygote hypercholesterolaemie in Nederland hoger dan eerder ingeschat
Literatuur - Sjouke B et al. Eur Heart J. 2014 - Eur Heart J. 2014 Feb 28
Homozygous autosomal dominant hypercholesterolaemia in the Netherlands: prevalence, genotype-phenotype relationship, and clinical outcome
Sjouke B, Kusters DM, Kindt I, et al.
Eur Heart J. 2014 Feb 28. [Epub ahead of print]
Achtergrond
Autosomaal dominante hypercholesterolaemie (ADH) kan worden veroorzaakt door mutaties in de genen die coderen voor de LDL-receptor (LDLR), apolipoproteïne B (APOB) of PCSK9. Homozygote ADH (hoADH) can het gevolg zijn van ofwel homozygotie of compound heterozygotie. hoADH wordt gekenmerkt door verhoogde niveaus van LDL-c en fysieke tekenen van cholesterolophopingen in de huid, ogen en/of pezen. De levenslange blootstelling aan hoge LDL-c niveaus leidt doorgaans tot cardiovasculaire aandoeningen (CVD) op jonge leeftijd in hoADH patiënten [1,2]. Statines verlagen de LDL-c niveaus in belangrijke mate, maar additionele behandeling, zoals bijvoorbeeld LDL-aferese, is vaak genoodzaakt in deze patiënten [3].De exacte prevalentie van hoADH is onduidelijk, deels omdat eerdere schattingen grotendeels gebaseerd waren op klinische, in plaats van moleculaire criteria [4]. Sinds de jaren ’90, is een cascade screeningprogramma in Nederland (NL) ingezet om alle ADH patiënten te identificeren. Omdat veel huisartsen en medisch specialisten zich bewust zijn van dit nationale programma, kan het gebruikt worden om de prevalentie en het klinische fenotype van moleculair vastgestelde hoADH in Nederland te onderzoeken.
Belangrijkste resultaten
- 104682 individuen werden gescreend op ADH mutaties. 49 patienten werden geïdentificeerd als drager van pathogene mutaties; 20 homozygoten (hoFH) en 25 compound heterozygoten (compHeFH) met mutaties in LDLR, en 4 homozygoten met APOB mutaties (hoFDB). Vier hoFH patients uit twee verschillende families hadden consanguine ouders.
- Gezien een populatie van 16722387 inwoners, varieert de prevalentie van hoFH en compHeFH in NL van 1 op 371608 (95%CI: 1: 287356 to 1: 526316) tot 1 op 407863 (95%CI: 1:312500 to 1: 588235) personen (na exclusie van nageslacht van consanguine ouders). De prevalentie van heterozygote FH (heFH) werd geschat op 1 op 319 personen.
- De prevalentie van hoFDB is 1 op 4180597 (95%CI: 2109705 to 1: 209205021), en heterozygote FDB wordt geschat op 1 op 1023 personen.
- Op basis van de berekende prevalenties, zijn er 68636 heterozygote ADH patiënten in NL, hetgeen een prevalentie van heterozygote ADH oplevert van 1 op 244 individuen (1/319 + 1/1023).
- Het soort mutatie beïnvloedt LDL-c niveaus in statine-naïeve patiënten: hogere LDL-c niveaus werden gezien in patiënten met een of twee null allelen, in vergelijking met patiënten zonder null allelen (17.7+2.6 vs. 9.1+2.9 mmol/L; P<0.001).
- 49% van de patiënten had LDL-c < 13.0 mmol/L, en voldeed dus niet aan de klinische criteria voor hoADH. Ongeveer 76% van de patiënten had niet LDL-c>7.8 mmol/L tijdens lipideverlagende therapie (een ander vaak gebruikt criterium).
- 30% van de hoADH patiënten maakte een CV event door, op een gemiddelde leeftijd van 34.2 +17.1 (range: 13-69 jaar).
- Alle patiënten van wie informatie over behandeling beschikbaar was, kregen lipideverlagende behandeling. Geen enkele patiënt behaalde het LDL-c doelwit zoals aanbevolen door de huidige ESC/EAS richtlijn (<2.5 mmol/L).
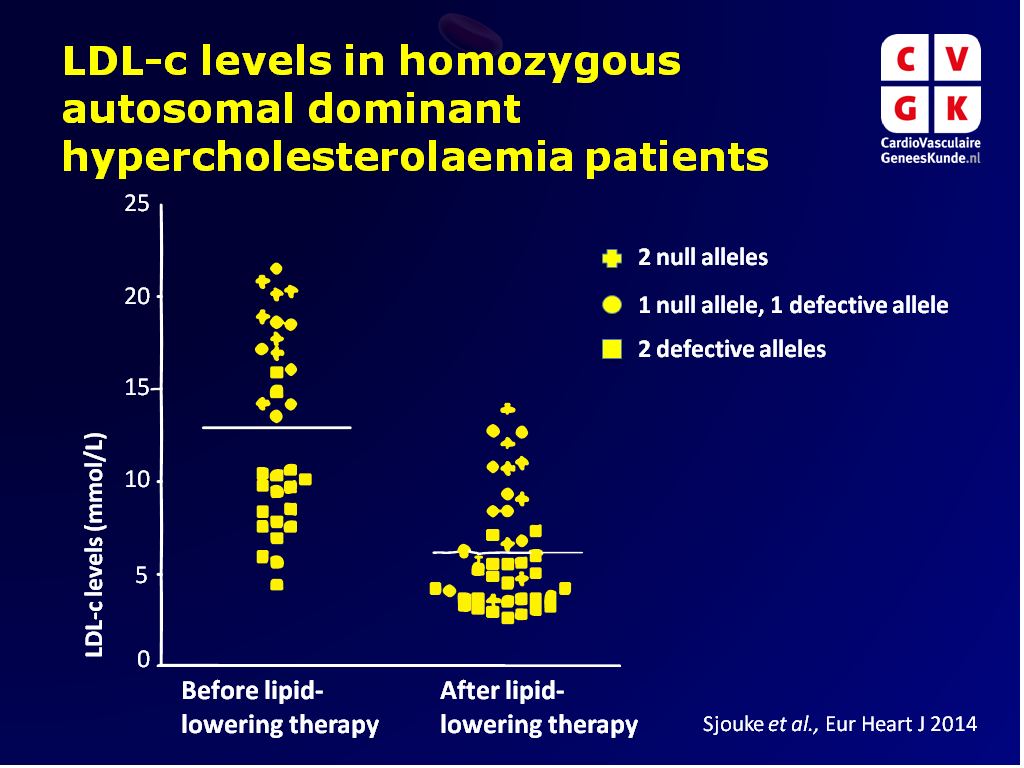
Conclusie
De prevalentie van moleculair vastgestelde hoADH (hoFH en compHeFH en hoFDB) in Nederland werd vastgesteld op ˜1 op de 300000, hetgeen ten minste drie keer zo vaak is als eerdere schattingen. Deze studie liet ook een aanzienlijke fenotypische variatie zien tussen patiënten met een moleculaire diagnose van hoADH, en, interessant genoeg, dat een groot deel van de patiënten niet voldeed aan de fenotypische criteria voor hoADH. Moleculaire diagnose van hoADH is belangrijk, omdat zowel de ouders als de kinderen van de hoADH patiënt heterozygote dragers zullen zijn. De huidige getallen suggereren dat totnogtoe slechts een derde van de heterozygote ADH gevallen is geïdentificeerd.Klik door naar dit artikel op Pubmed
Referenties
1. Goldstein JL, Hobbs HH, Brown MS. Familial hypercholesterolemia. In: Scriver C, Beaudet A, Sly W, Valle D (eds), The Metabolic and Molecular Bases of Inherited Disease. 8th ed. New York: McGraw-Hill; 2001. p. 2863 – 2913.
2. Kolansky DM, Cuchel M, Clark BJ,et al. Longitudinal evaluation and assessment of cardiovascular disease in patients with homozygous familial hypercholesterolemia. Am J Cardiol 2008;102:1438 – 1443.
3. Catapano AL, Reiner Z, DeBacker G, et al. ESC/EAS Guidelines for the management of dyslipidaemias The Task Force for the management of dyslipidaemias of the Euro- pean Society of Cardiology (ESC) and the European Atherosclerosis Society (EAS). Atherosclerosis 2011;217:3–46.
4. Raal FJ, Santos RD. Homozygous familial hypercholesterolemia: current perspectives on diagnosis and treatment. Atherosclerosis 2012;223:262 – 268.
Deel deze pagina met collega's en vrienden: